layout: page title: “Global Sequencing Fund” permalink: /sequencing-fund/
COVID-19 is global. Sequencing it should be too.
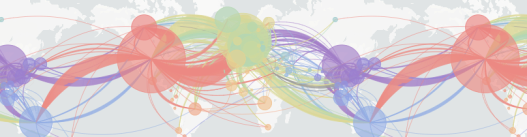
At Nextstrain, we depend on sequences generated by the hard work of labs around the world, who then share this data publicly on GISAID. The world has never seen so many sequences generated so rapidly and shared so quickly as now. However, there are still many countries and areas around the world for which we have no full-length, high quality sequences. A prime example is Iran - despite the devastating outbreak there, no full-genomes are available, seriously limiting local and global efforts to better understand how and when the outbreak may have started and how it spread both within and between other countries.
With the advent of relatively cheap and efficient sequencing techniques, like Oxford Nanopore’s minION range, sequencing is now more transportable, easily maintainable, and available, than ever before. However, a sequencer can only run with the appropriate reagents and limited-use ‘kit’ (which degrades over time and so must be replaced). Without these vital pieces, a lab can have the samples, expertise, and base sequencing equipment ready to start producing SARS-CoV-2 genomes - but stand unused for weeks or months.
The start of an epidemic is one of the most critical times to collect sequences: these unique dynamics are hard to capture after-the-fact, and countries need as much data as possible in order to make appropriate decisions about interventions such as social distancing and lockdowns. Many of sequencing centres are likely to be funded in future by local or governmental funding schemes, but will not be able to start processing critical samples now. Our aim is to quickly provide sufficient money to ensure that labs are not limited by reagents & kit, and can start generating and uploading sequences both for local response and for other scientists to use, as soon as possible.

In partnership with Fred Hutch, we’ve created an initiative to get labs enough money to cover the cost of reagents & kit to run about ~100 samples over the next few months. This is intended as ‘bridge’ funding to ensure sequences can be generated at this important time - during the growth and peak of the epidemic - while labs apply for longer-term funding.
Read more about the idea behind our initiative here
Donate here

How does sequencing help?
Sequencing allows us to see beyond simple case-counts. Through the viral genome, we can reconstruct how different viruses are related to each other (more on how we do this here) (link coming soon). This allows us to trace the virus as it moves through time and around the globe - without having to rely on patient interviews or self-reported movement or interactions (though we can often incorporate these too, if available).
As well as tracking the virus in real-time, we can make inferences about when and where the virus arrived in different parts of the world, country, or region - as data becomes more available. These efforts can be critical to inform appropriate health agency and government responses, both in the present and in the future.
While SARS-CoV-2 has a very normal mutation rate for a virus in the coronavirus family, and we have no concerns about the functional impact of viruses in the short-term, there is value in the long-term monitoring of the viral genetics. If SARS-CoV-2 becomes seasonal, and we develop vaccines and antiviral drugs to combat it, we will need to be able to monitor signs that the vaccines or treatment may be less effective, around the world and through time.
Globally
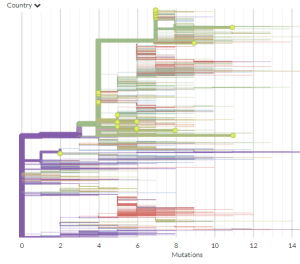
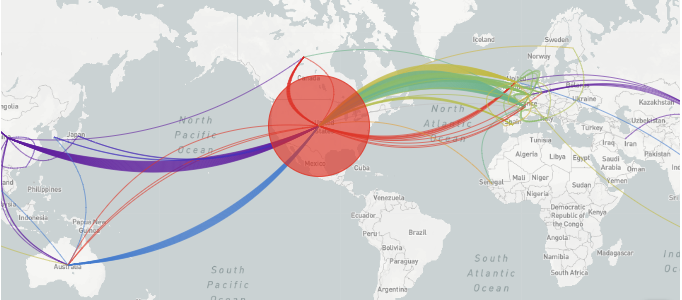
Sequencing helps us better understand how the virus is spreading between and within countries - without having to interview everyone with the virus. This helps show us, for example, that the epidemic in Italy was sparked by more than one introduction, and that the USA has had multiple introductions from all over the world (not ‘just’ from China - or Europe).
Locally
Sequencing can be critical at the beginning and end of an epidemic to figure out whether transmission is local or imported. For example, if we detected 5 cases in Townsville today, it’s really important to know whether these are from people who have arrived from ‘high risk’ areas - infected elsewhere and then arrived here - or whether these infections were transmitted right here in our community. Testing alone can struggle to distinguish these two, especially as sample numbers rise.
At the beginning of the epidemic, this important to decide what measures to take. If transmission is mostly being imported (the sequences are separate in the tree, and link to other countries or areas), then we might focus on what we can do to stop or isolate these imported cases. If transmission is mostly local (the sequences cluster really tightly in the tree), then we need to switch to measures like social distancing, isolation, increased testing, and possibly lockdowns - as we’ve seen in the past few weeks.
At the end of an epidemic, this same difference in imported or local cases can help us evaluate if our local containment measures are working. Cases might still be imported from other high-risk areas, despite our own local efforts, but as long as we can detect and isolate these, there’s no reason to modify our local interventions. However, if indications are that the cases due to local transmission, this might be an indicator that the measures that are implemented are not doing well enough - we may need to step up testing, or temporarily go back to lockdown measures.
This also allows for more targeted intervention - we can tell the difference between sequences being transmitted within one city or across the country - allowing us to take measures that just apply to the affected cities or regions, without needing to put restrictions back on the larger population - allowing social and economic recovery to continue on a larger scale, while we contain smaller areas where attention is needed.
Finally, by using the viral genetics to uncover how the virus has spread in the past, we can predict the routes it will use in the future. The transmission paths and transportation networks that were most conducive to aiding virus spread at the beginning of the outbreak are the same ones that will be susceptible to allowing transmission as we try to move out of social-distancing and lockdown restrictions. The better we understand how the virus moved through and between countries a few weeks and months ago, the better we can prepare to restrict transmission along these pathways in the future.
Long-term
As well as the immediate implications of sequencing for public health and government policy, there is a vital role for sequencing in longer-term surveillance, if the SARS-CoV-2 virus persists beyond this year.
Vaccine resistance
Work is already underway towards a SARS-CoV-2 vaccine. An effective vaccine will make a huge difference in how we can manage SARS-CoV-2 going forward, if the virus persists in the human population. However, we don’t know much about how immunity is formed and persists against viruses like SARS-CoV-2, nor what role mutations might play in the ongoing efficacy of such a vaccine. With influenza, we must update our vaccine twice a year (once each for the Northern and South hemispheres to prepare for their respective winters), due to mutations that occur on the outside of the virus. Other viruses do not mutate in the same way, and one vaccine is enough to protect you for the rest of your life. Where SARS-CoV-2 fits on this scale is unclear. What is clear, however, is that we must be prepared to monitor the virus for signs of these longer-term mutations, indications these may impact vaccines, and if necessary, use this information to select new vaccines.
Nextstrain, using sequencing data, plays an important role in helping to select these vaccine strains each year, and monitors the most important genetic sites in the influenza virus to give insight into how effective the old and new vaccines might be. Whether this role is filled by Nextstrain or by many platforms doing such tracking, we should be prepared now to start surveillance for these mutations in SARS-CoV-2.
Drug resistance
Similarly, as we develop antiviral drugs provide effective treatment against COVID-19 (the disease caused by SARS-CoV-2), we will need to monitor the SARS-CoV-2 virus for mutations that lead to drug resistance. This is already commonly done with HIV. Additionally, we may be able to assess whether different variants of the virus, which may arise in future, have differing susceptibility to antiviral treatments.
Read More & Donate
Read more about the idea behind our initiative here
Donate here